
White Paper
There is a common misconception that determining whether you need an unregulated Internet of Things (IoT) platform or a regulated IoT platform is about the product(s) hosted on the platform.
This is not the case, however. The need for a regulated platform is predicated on the intended use of the data collected and where the data are processed or analyzed, not about whether you’re hosting data from a regulated Class II or III (FDA) or Class IIa, IIb or Class III (EU) device.
In this white paper, we outline the limitations of an unregulated platform, or a platform for transferring data (known as a Medical Device Data System (MDDS) in the United States), including the types of use cases that an unregulated platform can and can not support.
We also discuss the importance of developing the regulatory strategy for your digital health products now, to be prepared for the future. Most MDDS platforms do not offer a sustainable regulatory infrastructure to support more mature digital health solutions.
From our code to our culture, BrightInsight is built to support regulated digital health solutions. We understand the highly regulated environment that our biopharma and medtech customers operate in, and as such, have built a team that’s committed to quality and a platform that is compliant with the standards and regulations needed by our customer’s various digital health solutions.
The purpose of this white paper is to clearly explain how the regulated BrightInsight Platform differs from an MDDS platform, and help you decide which type of infrastructure you need today and in the future.
There is substantial confusion around the digital health regulatory landscape.
Through our market research, interviews with regulatory thought leaders and conversations with customers in the field, it is apparent that global medical device software regulations are nuanced and that there are a number of complexities in the specific digital health use cases that biopharma and medtech companies are looking to deploy.
Within the evolution of digital health, an increasing number of progressive U.S. and European Medical Device regulations have come into effect. This forward-thinking positioning can cause a lot of uncertainty on how to qualify and validate digital health platforms and medical software. Biopharma and medtech executives usually don’t have the time to go through all this legal documentation and need support in navigating through the complex and constantly evolving regulatory maze.”
— Tanja Rohark, CEO & Founder, Digital Chameleon
The numerous updates on digital health from the FDA and other global authorities in recent years suggest that regulations for software products will evolve at an increasing pace, adding complexity to biopharma and medtech companies’ already demanding regulatory responsibilities. While authorities should be lauded for keeping up with the fast-moving digital health market by issuing new regulatory updates, keeping tabs on the current thinking has proven to be a regulatory challenge in its own right.
When it comes to the regulatory landscape for software used with drugs, you are layering uncertainty on uncertainty. There is uncertainty from the Center for Devices and Radiological Health (CDRH) and from the Center for Drug Evaluation and Research (CDER). The trend overall seems to be toward more regulation."
— Bradley Merrill Thompson, Epstein Becker & Green, P.C.
Biopharma and medtech execs also encounter a myriad of conflicting marketing materials and sales presentations from vendors attempting to blur the lines between underlying infrastructures that are “medical-grade” or ones that support “GxP”, the "good practice" quality guidelines. “GxP” is the general abbreviation where the “x” can stand for various fields, such as “GMP” / Good Manufacturing Practice or “GLP” / Good Laboratory Practice.
Having an infrastructure and the quality systems in place to show good practices for your manufacturing or IT systems is very different – and is regulated differently – than Software as a Medical Device.
Traditionally, biopharma companies only deal with regulated software when it comes to their IT systems involved in the drug development and manufacturing process and are typically limited to GxP. When we started our digital health journey we did not know what we did not know, and assumed we had the systems and the skills in place to support regulated digital health software development, but we did not."
— Senior digital health product executive formerly with a top 25 biopharma company
GxP requires validation of IT systems and not of the medical device software.
GxP is a term you increasingly encounter in both the biopharma and medtech industry and refers to guidelines for "good work practice". This is specifically important within software development as adhering to these guidelines promotes quality, such as having batch numbers or quality numbers for your drug or device. However, Digital Therapeutics or Software as a Medical Device (SaMD) are regulated differently than GxP, and the requirements vary greatly, including the way you conduct risk assessment, usability testing, clinical evaluation, validation and more. Our daily challenge is helping our customers understand these differences and to ensure their solutions are compliant and harmonized in both regulatory worlds."
— Anne Woitzik, Senior Manager Quality & Compliance, Digital Chameleon
Many biopharma and medtech executives grapple with whether or not to build solutions on top of a regulated or an unregulated software infrastructure. There are varying opinions on which strategy to take because there is no clear delineation, definition, or existing guidance document to make this decision any easier.
The first thing to understand is that comparing an unregulated platform, or a platform for transferring data (MDDS Platform) to the regulated BrightInsight Platform is like comparing apples to oranges.
Determining the need for an unregulated platform versus a regulated platform is predicated on the intended use of the data and if you’re analyzing the data on the platform, not about whether you’re hosting data from a regulated Class II or III (FDA) or Class IIa, IIb or Class III (EU) device.
To make this crystal clear, we’ve included some examples here:
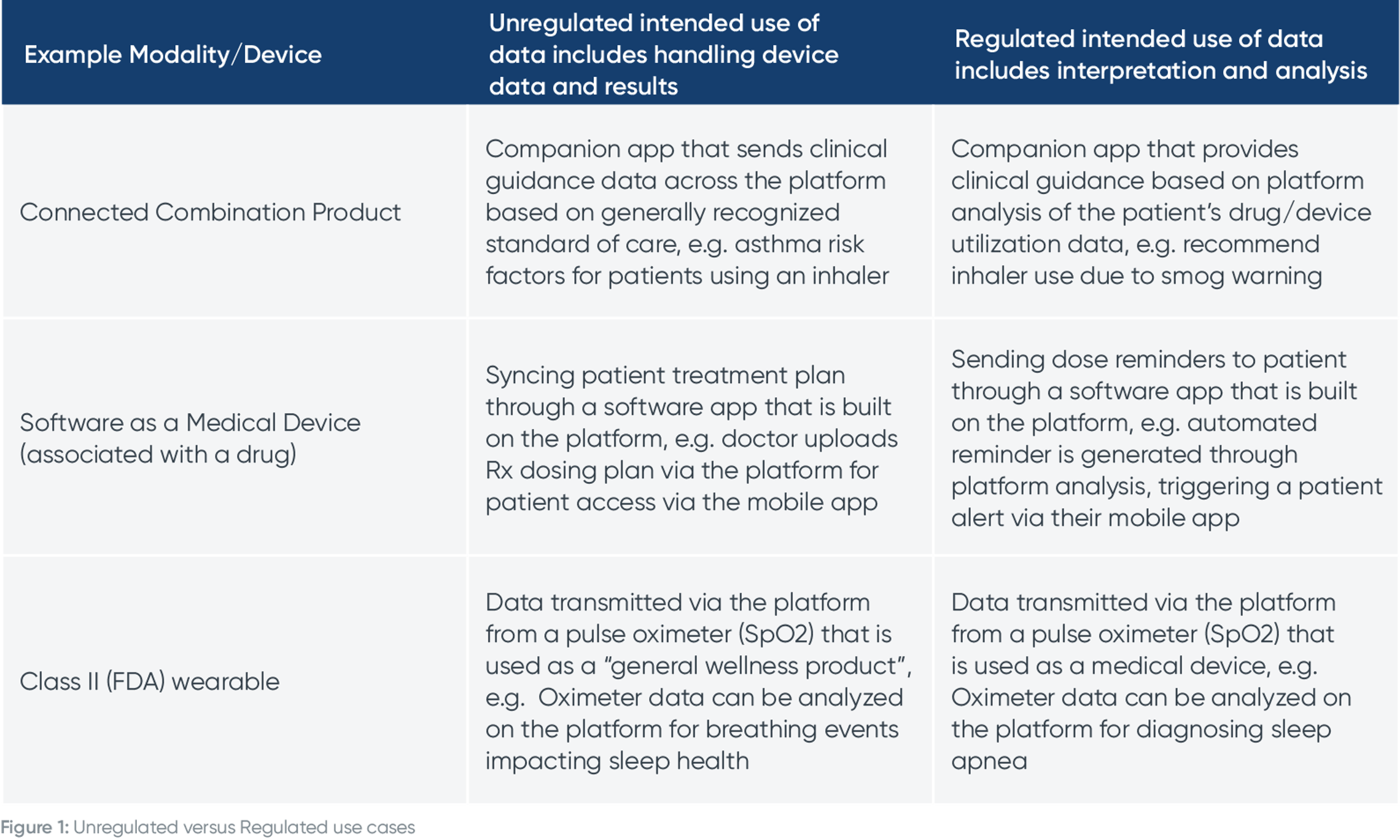
We have two other example use cases from a patient and physician viewpoint to crystalize this concept of regulated intended use of data.
Here’s an example of using data from a connected combination product in an unregulated versus a regulated way.

Here’s an example of using remote monitoring data in an unregulated versus a regulated way in a clinical trial.
Let’s say a patient is wearing a Class II (FDA) medical device that transmits data to caregivers. If a doctor were to review raw patient data and make a clinical decision about it, that is an unregulated use case. However, if you were to develop a Software as a Medical Device (SaMD) algorithm that analyzes data on the platform and makes clinical recommendations, that is a regulated use case.

Digital-savvy biopharma companies see adding new regulated functionalities as a way to improve patient engagement, deliver actionable insights to providers, and provide more value around their products and therapies.
Moving up the digital health regulatory maturity curve brings biopharma companies from developing simple companion apps, to capturing data from connected medical devices, to generating meaningful insights around the data, to building SaMD solutions. The more advanced regulated solutions include more robust feature sets, such as artificial pancreas systems, connected drug delivery devices, personalized drug dosing systems, and more.
Introducing more advanced capabilities like these can create operational efficiencies through automation and scale, improve patient outcomes through interventions and engagement, and ultimately optimize the value of connected drug, device or combination products. To unlock these benefits and deploy regulated digital health offerings, you need a strategy that includes a regulated IoT infrastructure.
Hopefully the earlier sections of this white paper make it clear that determining whether you need a regulated platform or an unregulated / MDDS platform is all about intended use of the data, not the regulatory status of the product(s) hosted on the platform.
As a biopharma or medtech company, it is important to understand that your intended use of the data will likely evolve over time, and you should plan for that.
Whether or not a biopharma or medtech company needs to use regulated software in their clinical trials is a complex question. We regularly encounter this precise challenge with our clients. One of the issues is determining if a software system used in a clinical trial does or doesn’t have a medical intent. A "medical intent" is the basis on which the classification of software as a medical device is determined. But—the regulatory authorities in the EU are becoming increasingly demanding and the scope of their interpretation of what is considered a medical device is expanding. Given this evolution, making the determination is not necessarily simple or straightforward."
– Elisabethann Wright, Partner, Hogan Lovells
You must contemplate your digital health solution roadmap and what type of regulatory strategy you need to support future products. MDDS platforms will not offer a sustainable regulatory infrastructure to support more mature digital health solutions.
Here’s an example for a connected inhaler, and how the intended use of the data changes over time:
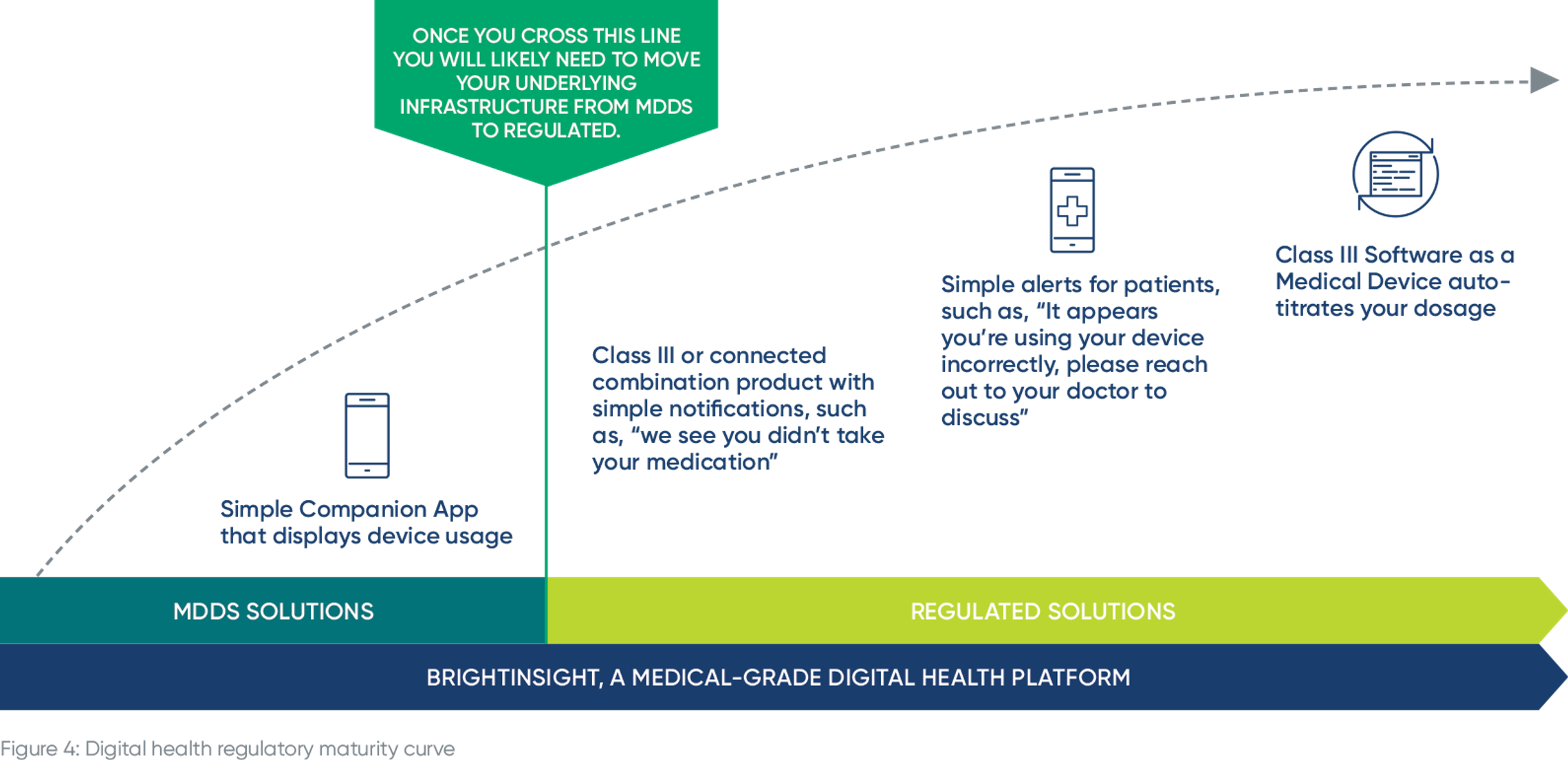
You may launch a simple companion app that displays inhaler usage. That’s not a regulated use case. But overtime, you want to improve patient adherence and outcomes so you deliver notifications and / or dosing recommendations based on an individual patient’s data analyzed on the platform. Once you make the leap from displaying inhaler usage to delivering dosing recommendations, that feature needs to be supported by a regulated infrastructure, with supporting documentation, quality system and more.
If your platform solution does anything beyond transferring, storing, converting the format or displaying medical device data – the definition of a Medical Device Data System (MDDS) – the chances are high that some aspect of a biopharma’s software will be regulated. If you want to manage risk, you need to adopt a more conservative point of view to future-proof your business."
– Bradley Merrill Thompson, Epstein Becker & Green, P.C.
Thinking even further down the line, you will likely want to be able to send alerts about using a product or therapy incorrectly, advising patients to reach out to their doctor, or even SaMD that can auto-titrate a patient’s inhaler dosage.
If you have an MDDS platform and you have a regulated intended use of the data on the platform, it might be possible to use your MDDS platform, but there’s a significant amount of remediation that needs to occur and that path is uncertain.
Let’s take the inhaler example from the previous section. You start with a simple companion app but decide you want to provide alerts to your patients about incorrect inhaler usage from SaMD on the platform. In order to introduce this functionality, you need to invest significant time and resources to secure approval on the regulated intended use of the data.
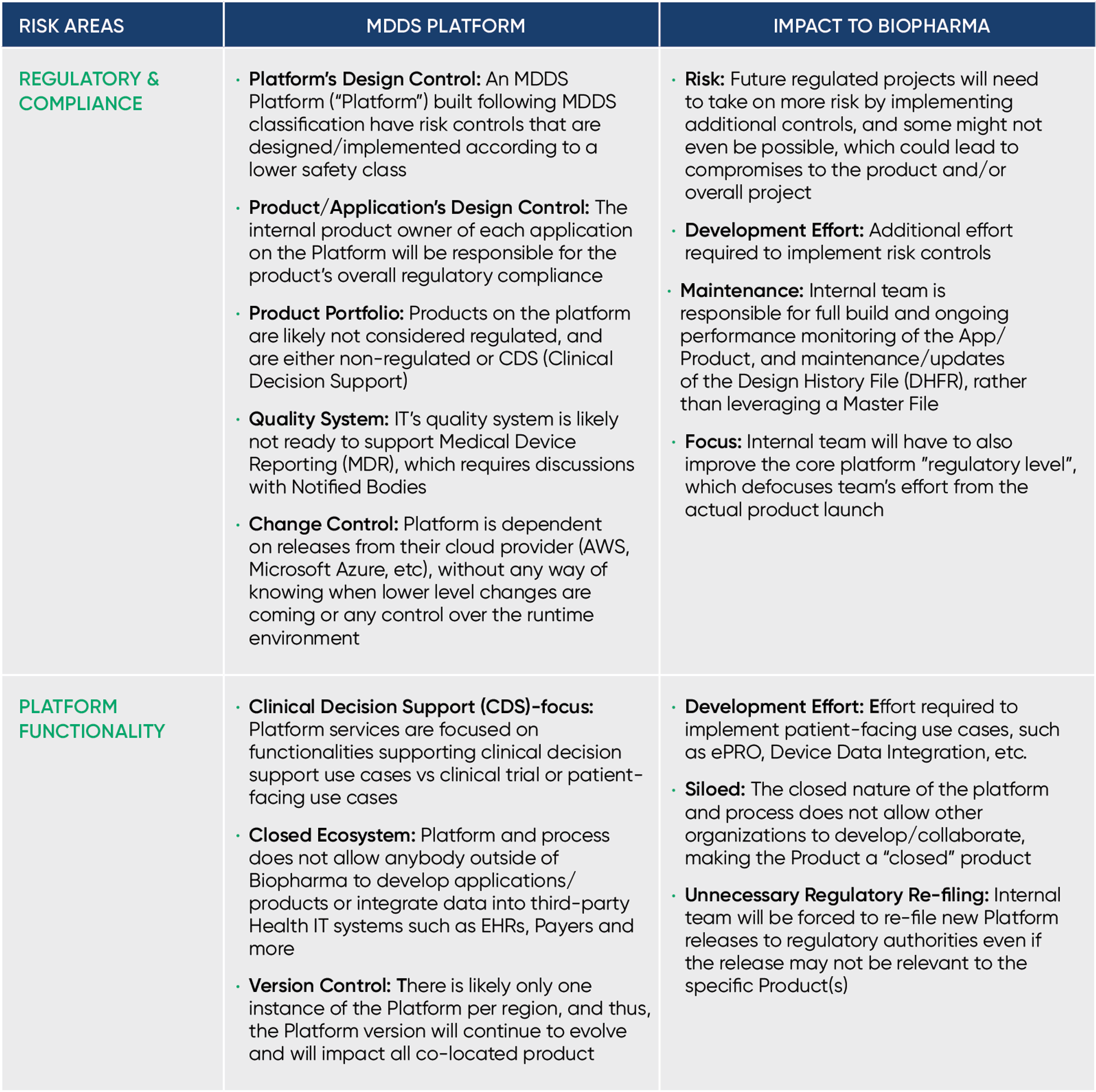
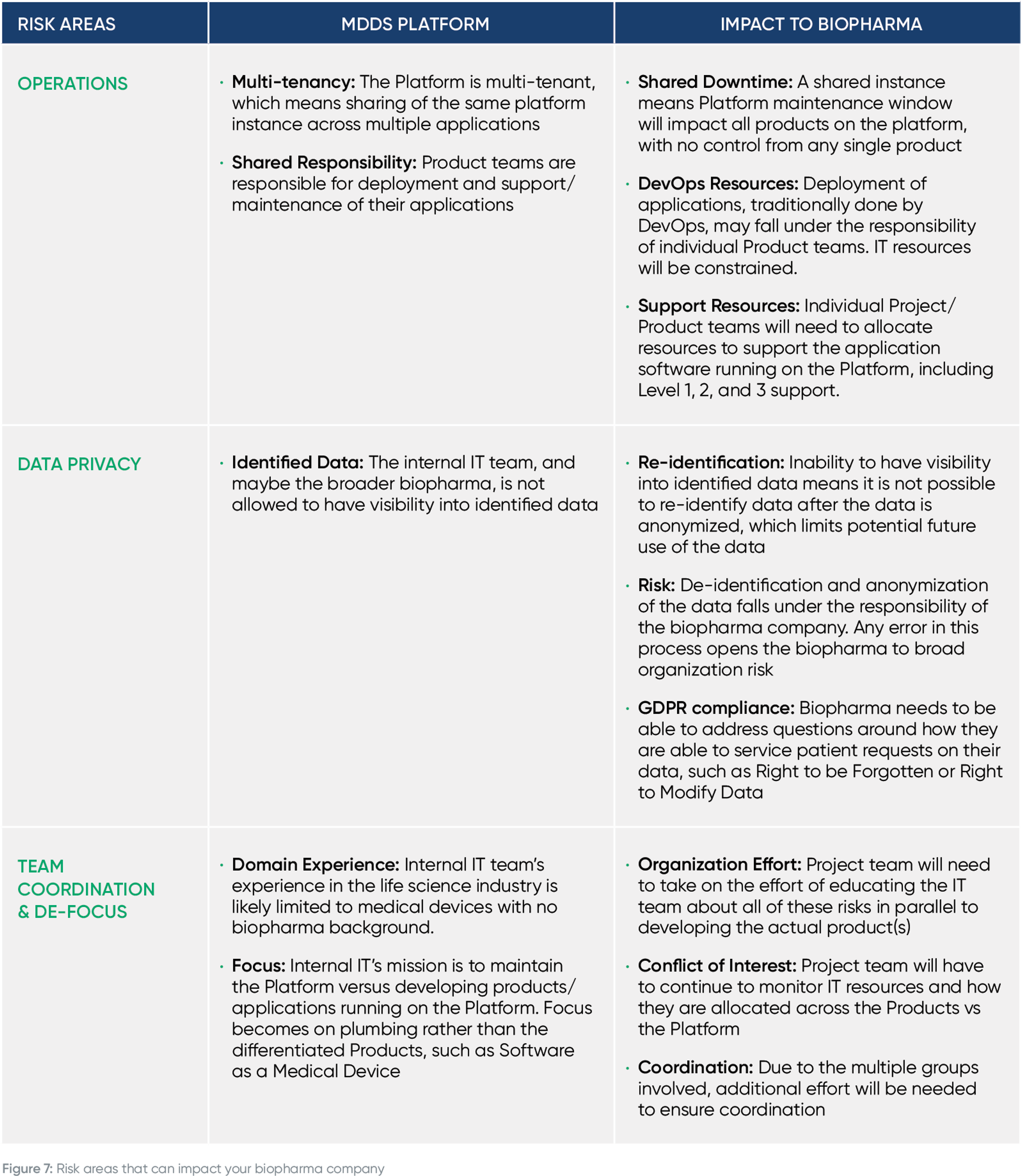
The below table displays the main ways the required remediation effort will impact your company, including delayed time to market, increased risk and more.

We started our digital health journey and assumed we had the systems and the skills in place to support regulated digital health software development. It was only a year later as we were approaching the first regulatory hurdle that we realized our QMS was misaligned with both US and EU medical device regulations. We then began the long journey to rebuild our QMS amounting to direct and indirect costs of millions of USD. Right before our product launch, another significant gap was discovered in our ability to support post launch activities, something that biopharma companies usually don't have to deal with as it relates to software. In total, our incorrect assumptions on regulatory, QMS and operations requirements for medical device software cost us more than a year of delays which could have been prevented by partnering with a company that had such expertise."
– Senior digital health product executive formerly with a top 25 biopharma company
We all know the devil is in the details, so let’s dive deeper into the FDA’s requirements to support up to Class III intended uses. Note that your platform will not be compliant with FDA regulations if you are missing these components.
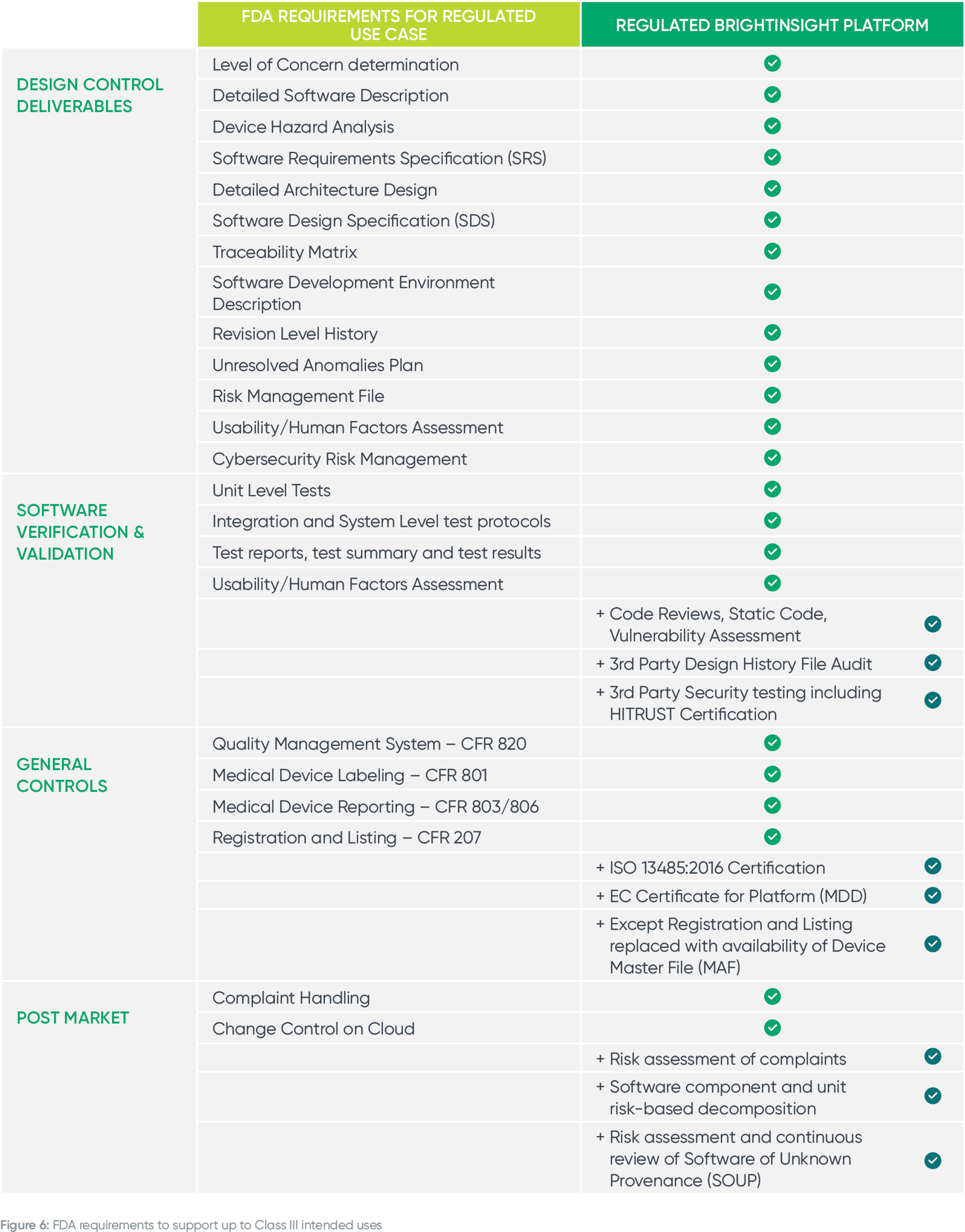
Building on the "Main Impact Areas" above, here are more details on how upgrading an unregulated or MDDS platform to support regulated use cases will delay your time to market, distract your team and increase compliance risk.
Having led the Regulatory and Quality programs for an MDDS platform in a prior role, I can confirm that the level of effort required to support a regulated use case is extensive. The work required from a Quality Management System and documentation standpoint alone requires an entire team, distracts from the core business and isn’t a sustainable strategy."
– Mark Tarby, Vice President, Regulatory and Quality Management System, BrightInsight
In our discussions with biopharma companies, we are regularly asked about whether or not they need a regulated IoT Platform for clinical trials. There is often a desire to use an unregulated platform that is homegrown for a low cost, quick way to capture data in a trial setting.
This is not a solid strategy, however, and will delay your time to market and increase your regulatory risk once you transition the product from clinical trials to commercialization.
If you use an unregulated platform when trialing a new therapy, combination product, companion app, or SaMD where data analysis takes place on the platform, you will run into issues when submitting your product to the FDA for approval. You will need to complete the remediation effort we discuss in the previous section of this white paper, which will delay time to market and introduces regulatory complexity and risk.
It’s important to use a regulated platform from the very beginning, starting with your clinical trials. Many biopharma execs believe that software regulations don’t apply when the software is used in clinical trials. Regardless of whether you choose to commercialize your Software as a Medical Device, you will have needed to document the design of the software from the beginning, before the trial even starts."
– Paul Upham, Head of Smart Devices, Roche/Genentech
Our customers who have trialed products on BrightInsight are grateful when it comes time to commercialize as all of the necessary documentation, quality systems, testing and more are in place for a seamless transition and product launch. If you use BrightInsight’s regulated infrastructure and QMS from the start of a trial, your regulatory approval will be much smoother as you will not have to make infrastructure or data transaction changes. The entire system (the IoT infrastructure and the clinical product) has already been validated within the clinical trial.
As the leading regulated IoT platform for biopharma and medtech, BrightInsight has achieved the utmost privacy, security, regulatory and quality certifications to minimize customer risk and protect sensitive health information. BrightInsight is built from the ground up to securely manage regulated medical device data and personal health information and is designed to support up to Class III medical device and combination product intended uses.
At Roche, most of our commercial products and clinical trials are multi-national, so our regulatory strategy needs to be contemplated across regions and across varying regulations. This will be the case for most leading biopharma companies. If you leverage a solution like BrightInsight that meets the most stringent requirements and maintains compliance as part of their managed service, you don’t have to worry about your regulated digital solutions in the U.S. versus Europe versus U.K. and so on. You just know they’re compliant."
– Paul Upham, Head of Smart Devices, Roche/Genentech
Biopharma and medtech companies wouldn’t accept a sub-par security, privacy, regulatory or quality system for their traditional drugs or devices, and the same rigor should be applied to their digital health offerings.
We are unquestionably moving towards a time where software used in clinical trials will require a regulated infrastructure in the EU. In the interest of caution, a regulated infrastructure is preferable because manufacturers can rely on this infrastructure to demonstrate the appropriate classification and, where necessary, the safety and effectiveness of their software solutions."
– Elisabethann Wright, Partner, Hogan Lovells
We are committed to the highest quality standards and BrightInsight, Inc. is EN ISO13485:2016 certified and our software development lifecycle process follows EN / IEC 62304. As part of our managed service, we maintain all of the required documentation and processes to ensure regulatory compliance globally.
From a security perspective, the BrightInsight Platform is HITRUST CSF® v9.1 Certified and HITRUST Certified of the NIST Cybersecurity Framework to manage risk, improve security posture and meet compliance requirements. The BrightInsight Platform also has certification for compliance with EN ISO 27001:2013. To support our commitment to the utmost privacy standards, the BrightInsight Platform is HIPAA and GDPR compliant and certified under both the EU-U.S. and Swiss-U.S. Privacy Shield frameworks. The BrightInsight Platform has also achieved the French HDS (“Hébergeur de Données de Santé”) certification, validating that BrightInsight ensures data confidentiality, integrity, and availability for our biopharma and medtech customers.